Once the molecule file is fully loaded, the image at right will become live. At that time the "activate 3-D" icon ![]() will disappear.
will disappear.
Sickle Cell Disease
The hemoglobin most commonly found in human adults is known as hemoglobin A. Hemoglobin A is composed of four subunits, two copies of hemoglobin α and
two of hemoglobin β. They are held together with salt bridges, hydrogen bonds, and hydrophobic interactions.
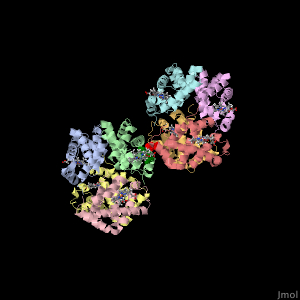
Sickle-cell disease is an inherited group of blood disorders that result from a mutated hemoglobin. The most common type is sickle-cell anemia. Sickle-cell anemia causes the two β subunits in hemoglobin A to be replaced with a different S subunit. The mutation in hemoglobin S replaces a single glutamic acid with valine at position 6 in the amino acid, which is shown as red spheres at the interface between two hemoglobin tetramers. The mutation to a hydrophobic amino acid allows it to interact with a phenylalanine and a leucine in the neighboring hemoglobin tetramer through the hydrophobic effect. This allows multiple mutated hemoglobin tetramers to hook up and form long strands that significantly reduces the cell's elasticity.
Sickle-cell disease is an inherited group of blood disorders that result from a mutated hemoglobin. The most common type is sickle-cell anemia. Sickle-cell anemia causes the two β subunits in hemoglobin A to be replaced with a different S subunit. The mutation in hemoglobin S replaces a single glutamic acid with valine at position 6 in the amino acid, which is shown as red spheres at the interface between two hemoglobin tetramers. The mutation to a hydrophobic amino acid allows it to interact with a phenylalanine and a leucine in the neighboring hemoglobin tetramer through the hydrophobic effect. This allows multiple mutated hemoglobin tetramers to hook up and form long strands that significantly reduces the cell's elasticity.