While proteins and nucleic acids consist of a limited number of building blocks, small molecules can contain a large variety of chemical groups, making accurate coverage for all possible compounds difficult. Although many general force fields such as CGenFF, GAFF and OPLS exist, coverage of all required parameter terms should be investigated before using such a force field. For example, ParamChem program offers automatic assignment of CGenFF atom types and parameters together with the penalties of those parameters. If these penalties are higher than 50, validation and reoptimization of the parameters are needed. In our group we develop accurate CHARMM-compatible parameters for small molecules, as needed, directly from ab initio calculations using the Force Field Toolkit (ffTK) plugin in VMD.
Validation
While agreement with ab initio data is important for force-field parameters, it is not sufficient for accurate parameter optimization. Therefore, we validate our developed parameters by checking the agreement with ab initio minimum geometries as well as making sure that the compound’s dynamics in simulations are realistic. The parameter optimization procedure is often iterative and repeated until sufficient agreement with both ab initio data, geometries, and dynamics is achieved.
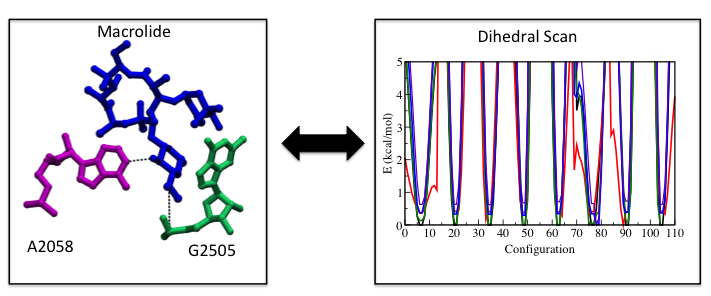
Our force-field parameters for macrolides were iterated in order to obtain more realistic dynamics of macrolides in the ribosome.
Applications
We recently used ffTK to develop accurate force field parameters for (1) macrolide antibiotics and (2) several forms of vitamin B12. The force-field parameters for macrolides were used to study several of them in wild-type and mutated, macrolide-resistant ribosomes. See for more on our research on macrolides.
Related publications
- Development of CHARMM-compatible force-field parameters for cobalamin and related cofactors from quantum mechanical calculations.
A. Pavlova, J. M. Parks, and J. C. Gumbart. J. Chemical Theory and Computation. 14:784-798, 2018. - Toward the rational design of macrolide antibiotics to combat resistance.
A. Pavlova, J. M. Parks, A. K. Oyelere, and J. C. Gumbart. Chemical Biology & Drug Design. 90:641-652, 2017. - Parametrization of macrolide antibiotics using the force field toolkit.
A. Pavlova and J. C. Gumbart. J. Computational Chemistry. 36:2052-2063, 2015. - Rapid parameterization of small molecules using the force field toolkit.
C. G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid, and J. C. Gumbart. J. Computational Chemistry, 34:2757-2770, 2013.