Group members attended all three of the major meetings this spring – BPS in San Francisco, APS in Denver, and ACS right here in Atlanta. All had great science to be sure, but I think ACS wins not just because of the great location but also because of the puppies they had on site!

#BPS2025

It’s hard to believe that another BPS has already come and gone! This year, a few of us went to LA for good food, better science, and, best of all, a chance to catch up with old friends! And now we’re already looking forward to San Francisco next year where we hope to see everyone again.
The next generation

The second generation of graduate students have completed their PhDs! Congratulations to Dr. Zijian Zhang, Dr. David Ryoo, Dr. Andrew Pang, and Dr. Katie Kuo for making it to the end! Or is it just the beginning? That’s for each of them to discover!
A reunion of sorts
 We just had our first *full* in-person gathering as a group in over two years! It was bittersweet however, as we got together to send off Atanu Acharya, who’s leaving to start his own lab in the Department of Chemistry at Syracuse University. Good luck to Atanu in his new career as an Assistant Professor – Syracuse will be lucky to have him!
We just had our first *full* in-person gathering as a group in over two years! It was bittersweet however, as we got together to send off Atanu Acharya, who’s leaving to start his own lab in the Department of Chemistry at Syracuse University. Good luck to Atanu in his new career as an Assistant Professor – Syracuse will be lucky to have him!

Over a year of coronavirus simulations
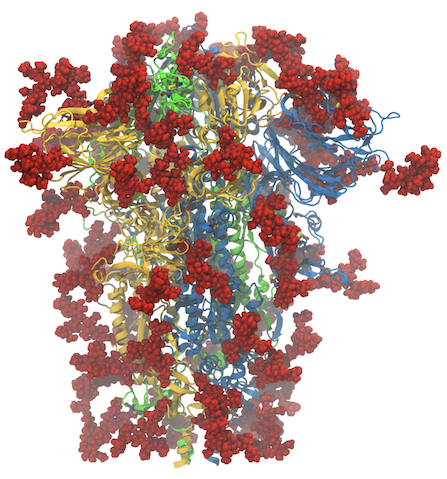 Like many computational labs, at the start of the pandemic in the US in early 2020, we turned our attention to SARS-CoV-2, aided by a rapidly growing amount of structural data. Since then, as a community, we have learned a tremendous amount about the biophysics of the virus and the proteins it uses to infect and replicate. Our lab in particular has focused on protonation states of the main protease (Mpro), the closed-to-open transition of the spike protein, and its binding to the receptor ACE2 (see our coronavirus page for more). The last year and a half have also demonstrated a tremendous degree of communication and cooperation amongst the scientific community and especially the computational chemistry community, including in two ACS symposia at the spring meeting (“A Call to Action: The Many Roles of Computational Chemistry in Addressing COVID-19” organized by JC Gumbart and Rommie Amaro) and the fall meeting (“Computational Chemistry of COVID-19: Lessons Learned and Future Directions” organized by Katarzyna Świderek and Carlos Simmerling). What discoveries will the next year and a half bring?
Like many computational labs, at the start of the pandemic in the US in early 2020, we turned our attention to SARS-CoV-2, aided by a rapidly growing amount of structural data. Since then, as a community, we have learned a tremendous amount about the biophysics of the virus and the proteins it uses to infect and replicate. Our lab in particular has focused on protonation states of the main protease (Mpro), the closed-to-open transition of the spike protein, and its binding to the receptor ACE2 (see our coronavirus page for more). The last year and a half have also demonstrated a tremendous degree of communication and cooperation amongst the scientific community and especially the computational chemistry community, including in two ACS symposia at the spring meeting (“A Call to Action: The Many Roles of Computational Chemistry in Addressing COVID-19” organized by JC Gumbart and Rommie Amaro) and the fall meeting (“Computational Chemistry of COVID-19: Lessons Learned and Future Directions” organized by Katarzyna Świderek and Carlos Simmerling). What discoveries will the next year and a half bring?