Coronaviruses are enveloped RNA viruses that are distinguished by the numerous spike proteins projecting from their surfaces. While a number of examples are known, the most recent example, SARS-CoV-2, is especially well known for being the causative agent of COVID-19 and of the 2020 pandemic. The viral lifecycle, including cellular entry, replication, assembly, and release opens up multiple avenues for investigation suitable for molecular dynamics simulation, a few of which are described below.
Main protease
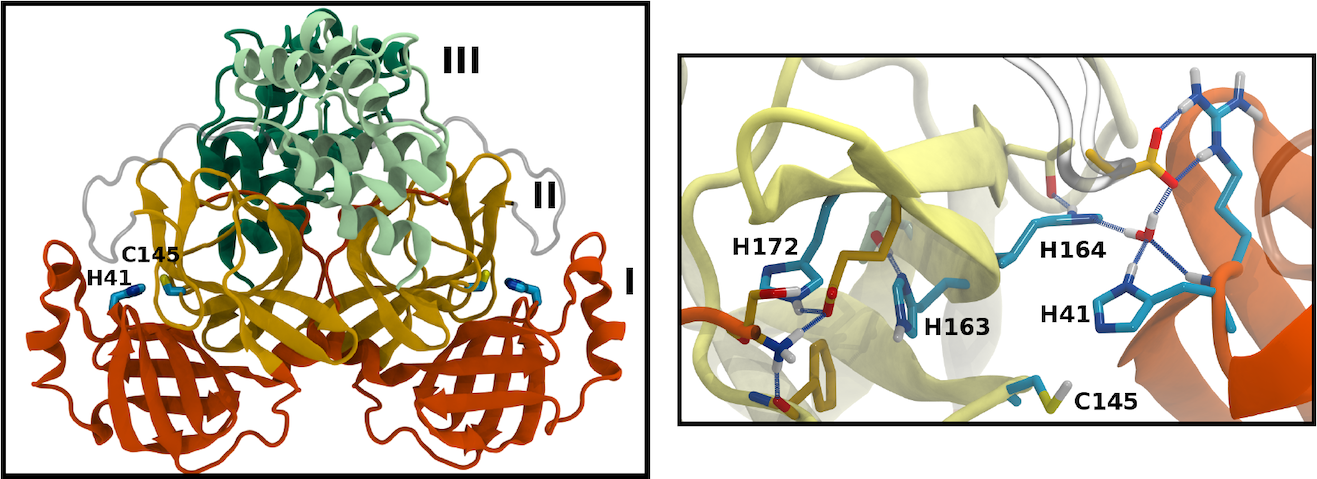
Many of the coronavirus proteins are synthesized together as a polyprotein, which is then cleaved at specific sites by viral proteases. One of these is the main protease (Mpro). The active site of Mpro includes a number of histidine residues, which can adopt one of multiple protonation states, normally unresolved by crystallography. We used molecular dynamics simulations to sample 12 different permutations of protonation states, judging them by the resulting stability of the protein and of the active site in particular.
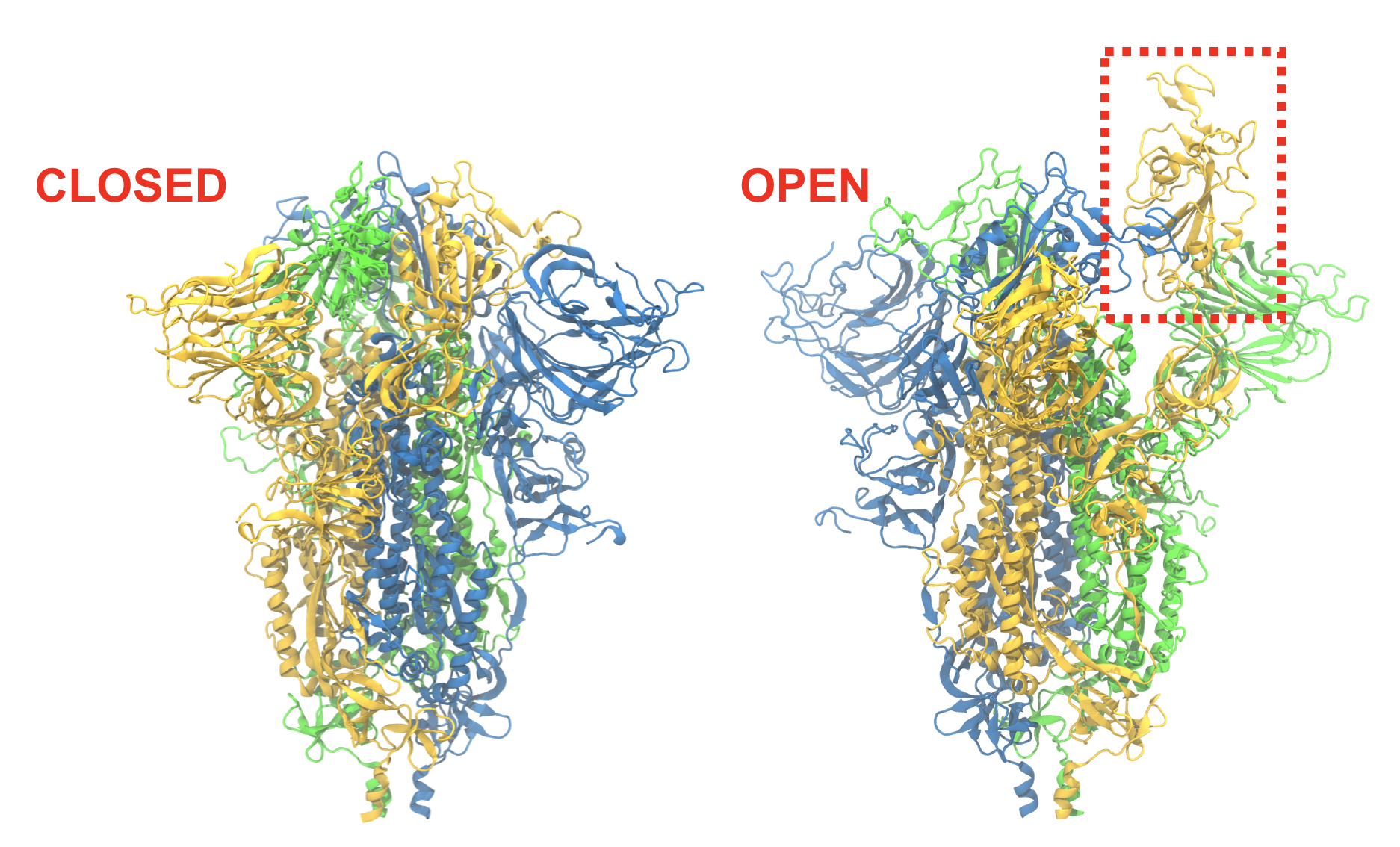
Spike conformational change
Before infection can take place, the receptor binding domain (RBD) on the spike protein must become accessible. The ease with which this closed-to-open transition takes place may determine in part how infectious the virus is. Free-energy calculations on the nation’s fastest supercomputer, Summit, will reveal the transition pathway as well as the energetics along it.
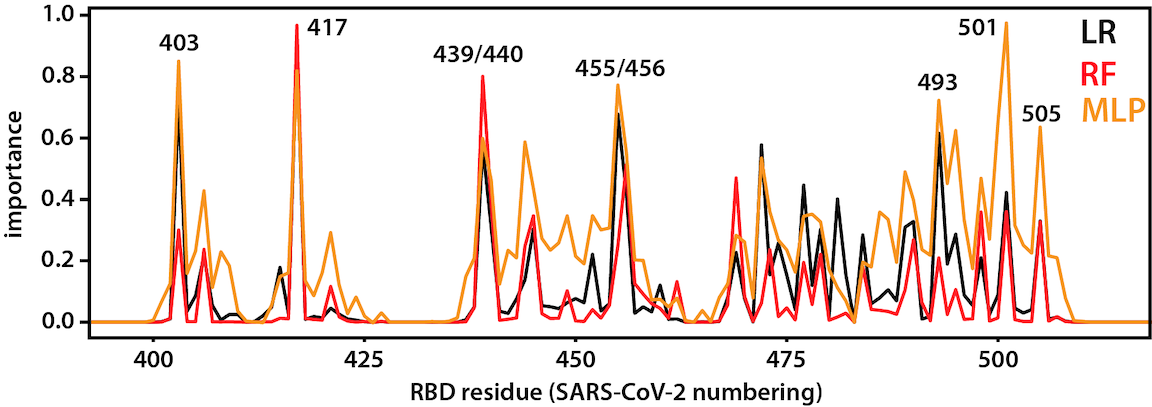
RBD-ACE2 interaction
After opening, the RBD binds to ACE2, a membrane protein on the surface of some human cells, notably lung alveolar epithelial cells. This happens to be the same binding site as that used SARS-CoV, a closely related virus that caused outbreaks in 2003. Machine-learning approaches applied to long trajectories of SARS-CoV-2 and SARS-CoV RBDs bound to ACE2 were used to identify which residues contribute to differences in binding. See here for the code used to do the analysis.
Recorded talk at UIUC (Nov. 2020)
Publications
- 9) Enhanced surface accessibility of SARS-CoV-2 Omicron spike protein due to an altered glycosylation profile.
D. Wang*†, Z. Zhang*, J. Baudys, C. Haynes, S.H. Osman, B. Zhou, J.R. Barr, and J.C. Gumbart†. ACS Infectious Diseases. 10:2032-2046, 2024. - 8) Species-agnostic polymeric formulations for inhalable messenger RNA delivery to the lung.
L. Rotolo*, D. Vanover*, N.C. Bruno*, H.E. Peck, C. Zurla, J. Murray, R.K. Noel, L. O’Farrell, M. Araínga, N. Orr-Burks, J.Y. Joo, L.C.S. Chaves, Y. Jung, J. Beyersdorf, S. Gumber, R. Guerrero-Ferreira, S. Cornejo, M. Thoresen, A.K. Olivier, K.M. Kuo, J.C. Gumbart, A.R. Woolums, F. Villinger, E.R. Lafontaine, R.J. Hogan, M.G. Finn, and P.J. Santangelo. Nature Materials. 22:369–379, 2023. - 7) SARS-CoV-2 spike opening dynamics and energetics reveal the individual roles of glycans and their collective impact.
Y.T. Pang*, A. Acharya*, D.L. Lynch, A. Pavlova, and J.C. Gumbart. Communications Biology. 5:1170, 2022. - 6) When the dust has settled: Calculation of binding affinities from first principles for SARS-CoV-2 variants with quantitative accuracy.
E. Goulard Coderc de Lacam*, M. Blazhynska*, H. Chen, J.C. Gumbart†, and C. Chipot†. J. Chemical Theory and Computation. 18:5890–5900, 2022. - 5) Machine Learning Reveals the Critical Interactions for SARS-CoV-2 Spike Protein Binding to ACE2.
A. Pavlova*, Z. Zhang*, A. Acharya, D.L. Lynch, Y.T. Pang, Z. Mou, J.M. Parks, C. Chipot†, and J.C. Gumbart†. J. Physical Chemistry Letters. 12:5494–5502, 2021. - 4) ACE2 glycans preferentially interact with SARS-CoV-2 over SARS-CoV.
A. Acharya†, D.L. Lynch, A. Pavlova, Y.T. Pang, and J.C. Gumbart†. Chemical Communications. 57:5949-5952, 2021. - 3) Tuning Proton Transfer Thermodynamics in SARS-CoV-2 Main Protease: Implications for Catalysis and Inhibitor Design.
L. Zanetti-Polzi, M.D. Smith, C. Chipot, J.C. Gumbart, D.L. Lynch, A. Pavlova, J.C. Smith, and I. Daidone. J. Physical Chemistry Letters. 12:4195-4202, 2021. - 2) Inhibitor binding influences the protonation states of histidines in SARS-CoV-2 main protease.
A. Pavlova, D.L. Lynch, I. Daidone, L. Zanetti-Polzi, M.D. Smith, C. Chipot, D.W. Kneller, A. Kovalevsky, L. Coates, A.A. Golosov, C.J. Dickson, C. Velez-Vega, J.S. Duca, J.V. Vermaas, Y.T. Pang, A. Acharya, J.M. Parks, J.C. Smith, and J.C. Gumbart. Chemical Science. 12:1513-1527, 2021. - 1) Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19.
A. Acharya, R. Agarwal, M. Baker, J. Baudry, D. Bhowmik, S. Boehm, K. Byler, L. Coates, S.Y.-C. Chen, C.J. Cooper, O. Demerdash, I. Daidone, J. Eblen, S.R. Ellingson, S. Forli, J. Glaser, J.C. Gumbart, J. Gunnels, O. Hernandez, S. Irle, J. Larkin, T.J. Lawrence, S. LeGrand, S.-H. Liu, J.C. Mitchell, G. Park, J.M. Parks, A. Pavlova, L. Petridis, D. Poole, L. Pouchard, A. Ramanathan, D. Rogers, D. Santos-Martins, A. Scheinberg, A. Sedova, S. Shen, J.C. Smith, M.D. Smith, C. Soto, A. Tsaris, M. Thavappiragasam, A.F. Tillack, J.V. Vermaas, V.Q. Vuong, J. Yin, S. Yoo, M. Zahran, L. Zanetti-Polzi. J. Chemical Information and Modeling. 60:5832–5852, 2020.