A protein’s function is directly linked to both its chemical nature and 3-dimensional structure. One of the onuses of molecular dynamics simulations is to correctly predict the latter. MD force fields are constantly updated and refined to better match experimentally observed structures, with particular emphasis on populations of various secondary structures as well as the range of disordered structures.
Our lab is interested in the comparing the free energy of secondary structure formation for various MD force fields in order to assess their accuracy in correctly producing experimental distributions of structure populations. Using umbrella sampling with replica exchange molecular dynamics (REMD-US), we are mapping the free energy landscape of small peptides to better understand how small secondary structure motifs form in solution, and then apply these methods to protein folding in vivo.
α-helices
α-helices are some of the most common secondary structures found in native protein folds. Understanding the pathway of their formation and their stability in biological processes is necessary step in combating diseases associated with protein misfolding.
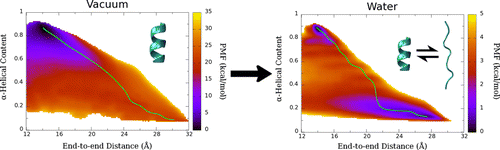
Potential of mean force (PMF) calculated for deca-alanine in water and vacuum. Green line shows the least free energy path calculated between the helical and extended states.
We examined the formation α-helix formation using the model peptide, deca-alanine. With many studies using deca-alanine in vacuum as a benchmark for free energy studies, we decided to examine the free energy landscape in explicit water. Although in vacuum, using the end-to-end distance of the peptide was sufficient to capture the important dynamics of the folding process, a second reaction coordinate, α-helical content, was necessary in order to fully characterize the folding process in water. What we observed was a new energy minimum in the extended conformations not seen in vacuum. These results were consistent with the NMR data used to optimized the CHARMM36 force field (used for these simulations), which revealed the PPII structure to be more prevalent in solution than the helix.
We noticed an overall reduction in energy from the vacuum to the solvated system, corresponding to hydrogen bonding with the surrounding water molecules. The extended, non-helical minimum also corresponds with an increase in peptide-water hydrogen bonds during the unfolding process. We ran additional equilibrium simulations to confirm our free energy surface. We were also able to map the equilibrium dynamics to the free energy surface for both the folding and unfolding processes (see below). We observed cooperative folding from the N-terminus towards the C-terminus, and unfolding from the C-terminus towards the N-terminus.
Equilibrium trajectory (right) mapped to the free energy surface (left), starting from an extended, non-helical state. High quality video here.
Equilibrium trajectory (right) mapped to the free energy surface (left), starting from the helical state. High quality video here.
β-hairpins
β-sheets are the other most common secondary structures found in native protein folds. Like α-helices, their structure arises through the formation of hydrogen bonds along the protein’s backbone.
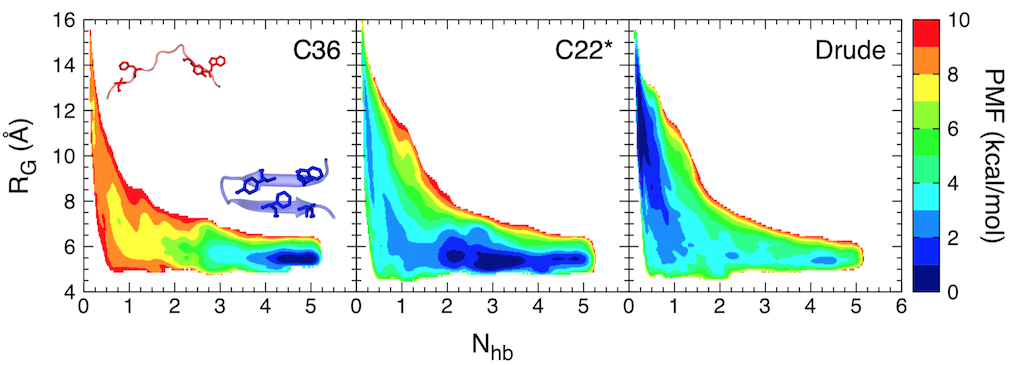
Free-energy landscape for folding of GB1 as a function of the number of native backbone hydrogen bonds (Nhb) and the radius of gyration (RG).
In this study, we focused on the C-terminal β-hairpin of the B1 domain of streptococcal protein G (GB1). Using replica-exchange umbrella sampling simulations, we determined the folding free energy landscape for GB1 using two fixed-charge CHARMM force fields, CHARMM36 and CHARMM22*, as well as a polarizable force field, the CHARMM Drude-2013 model, which has previously been shown to improve the folding properties of α-helical peptides. The CHARMM22* and Drude-2013 models are in rough agreement with experimental studies of GB1 folding, while CHARMM36 overstabilizes the β-hairpin.
Related publications
- Transmembrane but not soluble helices fold inside the ribosome tunnel.
M. Bañó-Polo, C. Baeza-Delgado, S. Tamborero, A. Hazel, B. Grau, I. Nilsson, P. Whitley, J. C. Gumbart, G. von Heijne, and I Mingarro. Nature Communications. 9:5246, 2018. - Folding free energy landscapes of β-sheets with non-polarizable and polarizable CHARMM force fields.
A. Hazel, E. Walters, C. Rowley, and J. C. Gumbart. J. Chemical Physics. 149:072317, 2018. - Structure and misfolding of the flexible tripartite coiled coil domain of glaucoma-associated myocilin.
S. E. Hill, E. Nguyen, R. K. Donegan, A. C. Patterson-Orazem, A. Hazel, J. C. Gumbart, and R. L. Lieberman. Structure. 25:1697-1707, 2017. - Thermodynamics of deca-alanine folding in water.
A. Hazel, C. Chipot, and J. C. Gumbart. J. Chemical Theory and Computation. 10:2836-2844, 2014. - Free energy of nascent-chain folding in the translocon.
J. Gumbart, C. Chipot, and K. Schulten. J. American Chemical Society, 133:7602-7607, 2011. - Diverse protein-folding pathways and functions of β-hairpins and β-sheets.
C. Balusek, H. Hwang, A. Hazel, K. Lundquist, A. Pavlova, and J. C. Gumbart. In L. Olivares-Quiroz, O. Guzman-Lopez, H.E. Jardon-Valadez, editors, Quantitative Models for Microscopic to Macroscopic Biological Macromolecules and Tissues. Springer. pp. 1-20. 2018.